
- Title
-
A versatile, multi-laser twin-microscope system for light-sheet imaging
- Authors
- Keomanee-Dizon, K., Fraser, S.E., Truong, T.V.
- Source
- Full text @ Rev. Sci. Instrum.
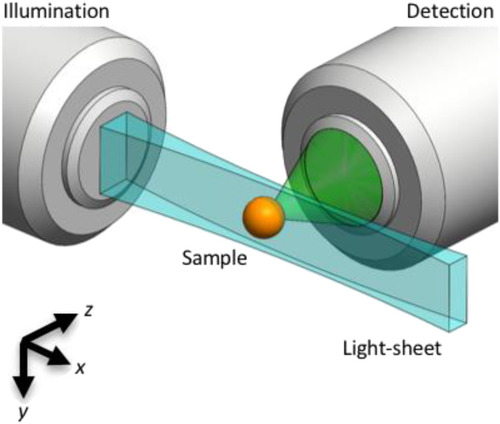
Light-sheet microscopy principle. A light-sheet (blue) can be created by dynamically scanning, along the y direction, a focused Gaussian beam that propagates in the |
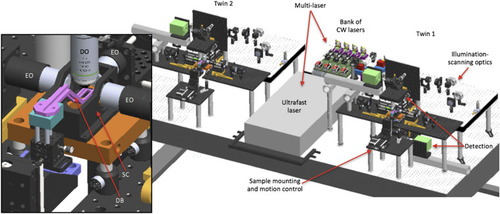
3D opto-mechanical model of the twin-microscope system mounted on a 5 × 10 ft2, anti-vibration optical table. Model shows the multi-laser subsystem shared between microscope-twin-1 (right) and microscope-twin-2 (left). Twin-1 has the four functional subsystems labeled and features an implementation of both upright and inverted detection. Brief descriptions of each functional subsystem are provided in |
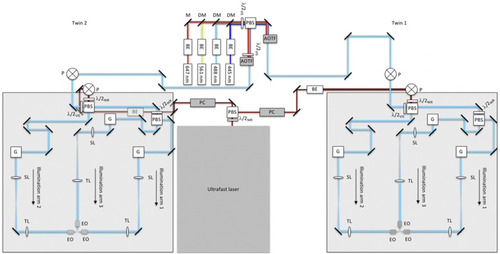
Schematic diagram of multi-laser and illumination-scanning optics subsystems of the instrument. Visible light from the continuous-wave laser bank is fed into microscope-twin 1 (right) and microscope-twin 2 (left) via polarization beamsplitting optics [consisting of a polarizing beamsplitter (PBS) and half-wave plate]. Acousto-optic tunable filters (AOTFs) are used to select the visible wavelengths and adjust the power independently for each twin. The near-infrared (NIR) light from the ultrafast laser is routed similarly using Pockels cells (PCs) to adjust the NIR power independently for each twin. The visible and NIR beams are raised onto 24 × 36 in.2 optical breadboards by using periscopes (P). Polarization beamsplitting optics are used both to combine the visible and NIR beams and to split the combined beam into two paths (illumination arms 1 and 2). Illumination arm 1 is further split into two paths through polarization beamsplitting optics, creating a total of three illumination arms. Each illumination arm directs light to the sample through the excitation objectives (EO). BE—beam expander, M—mirror, DM—dichroic mirror, λ/2—half-wave plate, where the subscripts VIS and NIR refer to the visible and near-infrared wavelengths, respectively, G—2D scanning galvo mirrors, SL—scan lens, and TL—tube lens. BE in the NIR twin-2 path appears gray because it is underneath the optical breadboard. |

Photograph of an assembled microscope-twin with upright detection. SC—sample chamber, SS—3D stage stack-up, DO—detection objective, FW—filter wheel, TL—tube lens, and CAM—camera. |
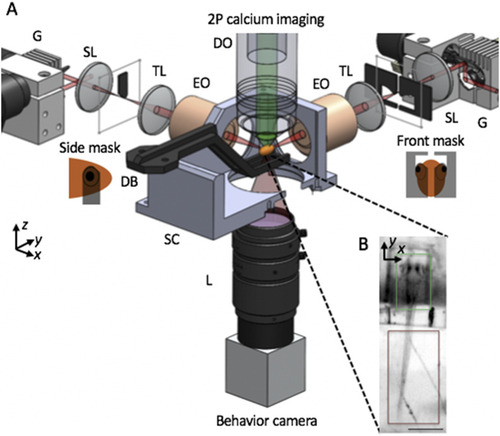
SPIM imaging of intracellular calcium for capturing neuronal activity. (a) Schematic of apparatus for imaging of neural activity during various behaviors in the larval zebrafish. Sheets of laser light are synthesized by quickly scanning the pulsed illumination beam (red) with galvo mirrors (G). 2P light-sheets are delivered to the agarose-embedded head of the animal with excitation objectives (EO) from the side and front arms. The side masks cover each eye on the sides of a horizontally oriented zebrafish, while the front mask covers both eyes, enabling access to neurons between the eyes. 2P-excited calcium fluorescence signal is collected through an upright detection objective (DO) and onto a scientific CMOS camera. A triggerable wide-field camera is positioned below the sample chamber (SC) to provide a wide-field, low-resolution view of the sample, as shown in (b). During a typical neural imaging experiment, the zebrafish larva is mounted in a caddy, which in turn is mounted to the dive bar (DB) underneath the DO. Within the caddy, the zebrafish’s head is immobilized in agarose, while the tail is free, permitting the monitoring of zebrafish behavior through tail movement. SL—scan lens, TL—tube lens, SC—sample chamber, and L—camera lens. The third illumination arm, emission filter, detection TL, scientific camera, light-emitting diode, and filter for behavior channel are not shown. Insets in (b) highlight that the calcium fluorescence channel (green) is recorded from the zebrafish brain, while the behavioral channel (dark red) monitors the tail movement of the animal. Scale bar: (b) 400 |
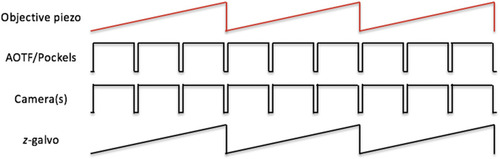
Schematic of control signal sequences for objective-scanning mode. The analog signal representing the position of the objective piezo collar is used as the master timing signal to generate control signals for the imaging cameras (both the fluorescence camera and behavior camera). The timing output of the fluorescence camera controls the AOTF/Pockels. The number of pulses driving the cameras, shown as 3 in the schematic here, determines the number of individual |
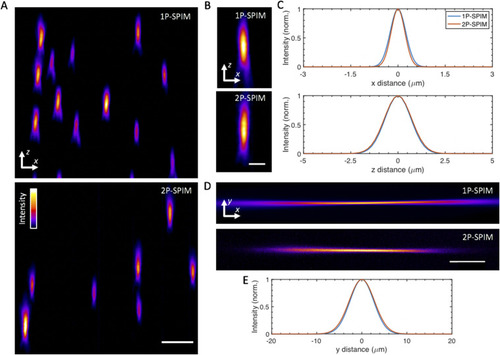
System imaging performance and characterization. (a) |
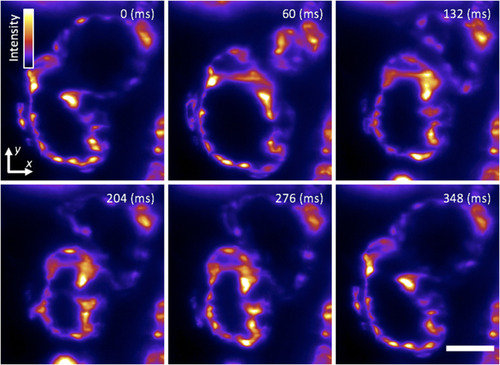
Cardiac light-sheet imaging. Single-plane SPIM recording of the beating heart in a live 5-dpf larval zebrafish with the endocardium fluorescently labeled (GFP), showing six distinct time points during the cardiac beating cycle. These subcellular 2D images are comparable to our previous efforts |
Light-sheet imaging of the dynamic motion of the beating heart of a 5-dpf transgenic larval zebrafish. Same dataset as presented in |
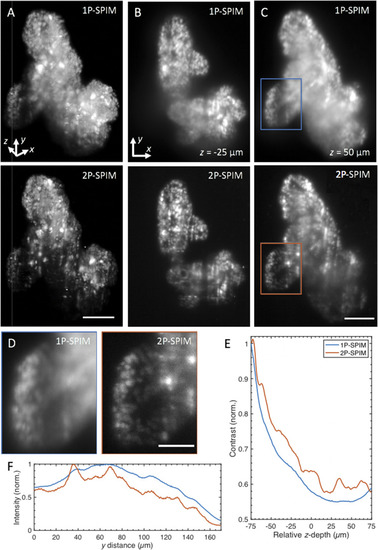
1P- and 2P-SPIM imaging of thick tumor organoids derived from a patient with colorectal cancer. (a) Volume rendering of fixed patient-derived tumor organoids expressing nuclear-localized H2B-GFP recorded in 1P (top) and 2P mode (bottom). Renderings show that the reduced background of 2P-SPIM enables better contrast throughout the imaged volume compared to 1P-SPIM. 3D organoid volume of ∼400 × 550 × 150 ( |
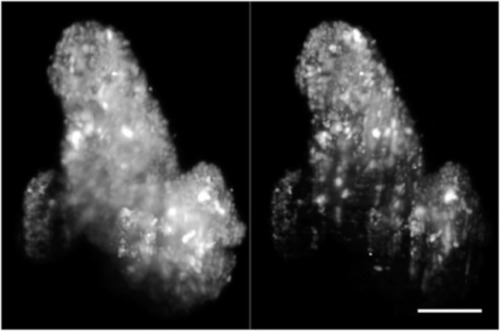
Volume rendering of fixed patient-derived tumor organoids expressing H2B-GFP, comparing images taken with one-photon (1P-, left) and two-photon excitation SPIM (2P-, right). Volumes are rotated around the |
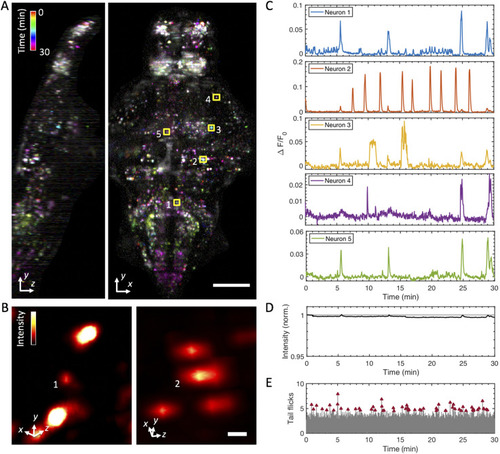
Whole-brain functional imaging at single-cell resolution in behaving 5-dpf transgenic larval zebrafish expressing nuclear-localized calcium indicator elavl3:H2B-GCaMP6s. (a) Maximum-intensity projections of calcium activity are color-coded in time over the 30-min recording window. Active neurons that exhibit fluorescence change during the recording appear as colored dots. Volume of 400 × 800 × 250 ( |
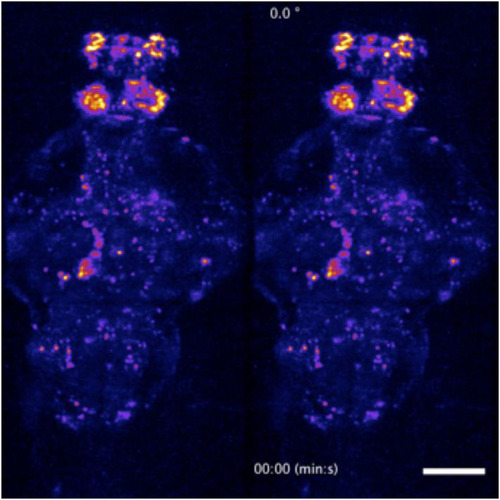
Dorsoventral (left) and rotating (right) maximum-intensity projections of a time-lapse recording of the whole-brain of the a 5-dpf transgenic larval zebrafish. Two-photon whole-brain functional light-sheet imaging was performed at a volumetric rate of 0.5 Hz. The video loops a 5-min recording as part of the data presented in |