
- Title
-
Precise in vivo functional analysis of DNA variants with base editing using ACEofBASEs target prediction
- Authors
- Cornean, A., Gierten, J., Welz, B., Mateo, J.L., Thumberger, T., Wittbrodt, J.
- Source
- Full text @ Elife
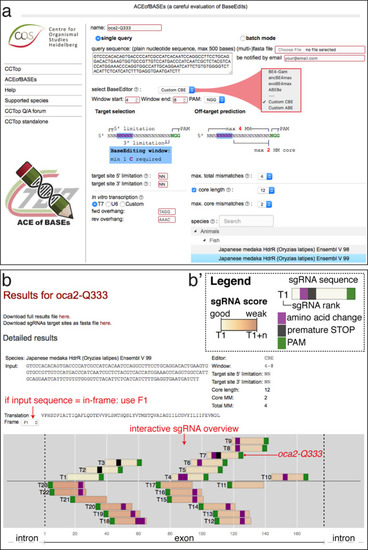
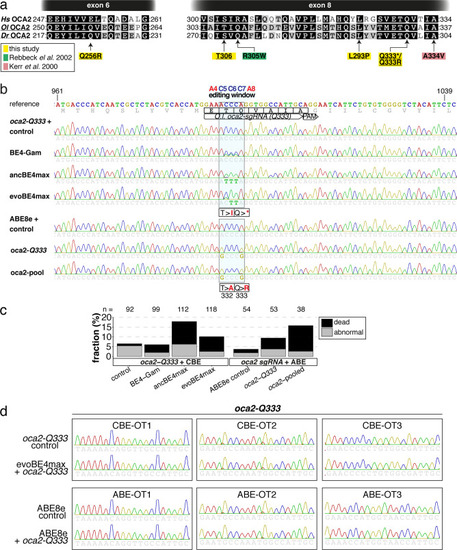
(a) User interface of the ACEofBASEs base editing design tool with base editor choice dropdown menu and a compendium of model species to select from. (b) Results page of the ACEofBASEs design tool for cytosine base editing of the Oryzias latipes oca2 locus. Using an in-frame sequence of the target site will directly provide the translation frame F1. Alternatively, frames can be selected from the dropdown menu. All sgRNA target sites found in the query are shown with potential amino acid change (magenta box) or nonsense mutation (PTCs, black box); here: standard editing window: nucleotides 4–8 on the protospacer; PAM: positions 21–23. For off-target prediction, a comprehensive list of potential off-target sites that contain an A or C in the respective base editing window is provided per sgRNA target. Potential off-target sites are sorted according to a position-weighted likelihood to introduce an off-target, that is the closer a mismatch at the potential off-target site to the PAM, the more unlikely this site is falsely edited (Stemmer et al., 2015).
|
( |
( |
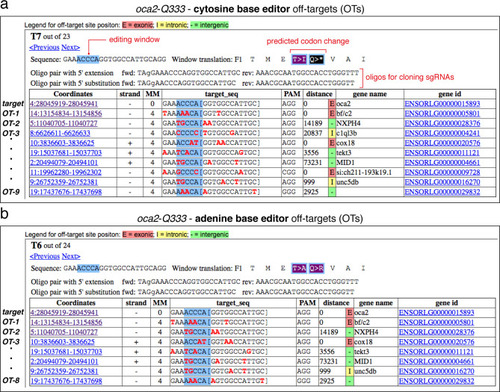
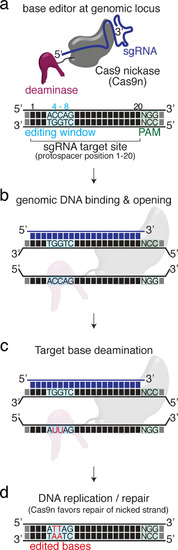
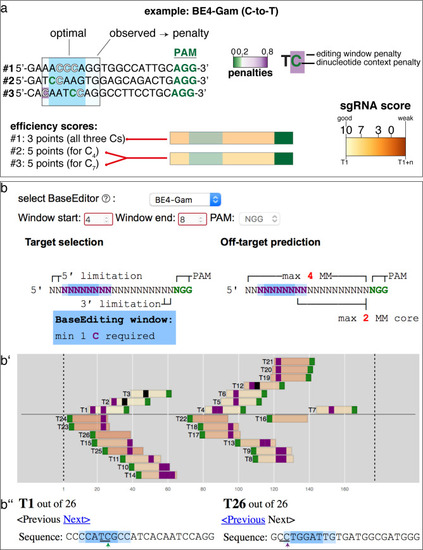
Overview of sgRNA |
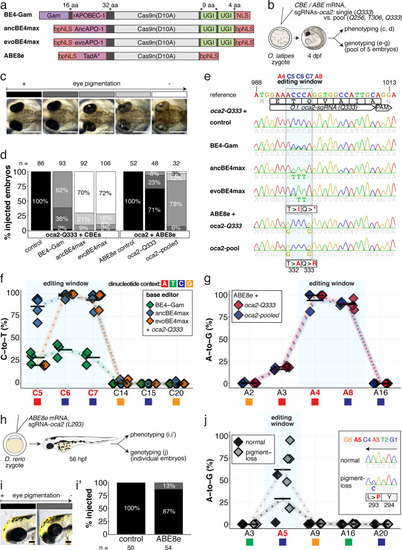
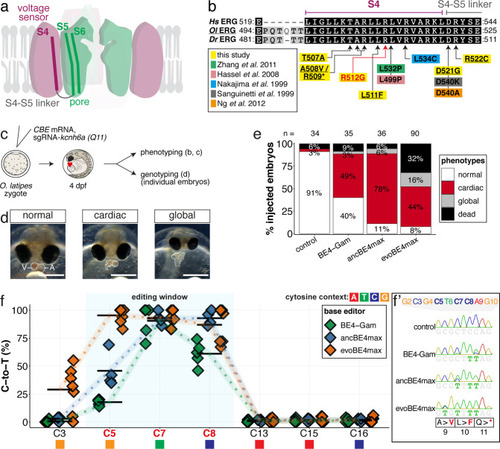
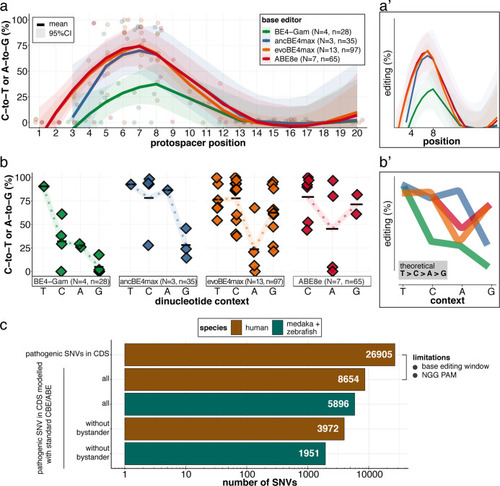
(a) Schematic diagram of the cytosine base editors BE4-Gam, ancBE4max and evoBE4max (evoAPOBEC1-BE4max) and the adenine base editor ABE8e. Cas9n-D10A nickase (light grey) with N-terminally linked cytidine or deoxyadenosine deaminase (pink) and C-terminal SV40 or bipartite (bp) nuclear localization sequence (NLS, red). All except BE4-Gam also contain the bpNLS N-terminally. CBEs contain variations of the rat APOBEC-1 cytidine deaminase, whereas ABE8e contains the TadA* domain (tRNA adenine deaminase), CBEs further contain C-terminally linked Uracil glycosylase inhibitors (UGI, green). Gam protein from bacteriophage Mu (purple) and linkers of varying lengths (dark grey). (b) Scheme of the experimental workflow. Cytosine or adenine base editor (CBE/ABE) mRNA and oca2-Q333 or a pool of three oca2-sgRNAs (–Q256, –T306, –Q333) were injected into the cell of a medaka zygote. Control injections only contained oca2-Q333 or ABE8e mRNA. (c) Phenotypic inspection of eye pigmentation was performed at 4 dpf (dorsal view). (d) Grouped and quantified pigmentation phenotypes shown for BE4-Gam, ancBE4max, evoBE4max, and ABE8e experiments. Control only contains oca2-Q333 sgRNA. n shown excludes embryos that are otherwise abnormal or dead, with abnormality rate given in supplement 1c. (e) Exemplary Sanger sequencing reads for each experimental condition, obtained from a pool of five randomly selected embryos at the oca2-Q333 locus. (f–g) Quantification of Sanger sequencing reads (by EditR, Kluesner et al., 2018) for BE4-Gam (n = 3), ancBE4max (n = 5) and evoBE4max (n = 3) (f), and ABE8e for single (n = 3) and pooled oca2-sgRNA experiments (n = 3) (g). Pools of five embryos per data point summarizes editing efficiencies. Mean data points are summarized in Supplementary files 1 and 2. To highlight the dinucleotide context, the nucleotide preceding the target C or A is shown by red (A), green (T), blue (C) and yellow (G) squares below the respective C or A. (h) Microinjections into the yolk of one-cell stage zebrafish were performed with ABE8e mRNA and oca2-L293 sgRNA. Zebrafish larvae were phenotypically analyzed at 56 hpf and individual larvae were subsequently genotyped. (i-i’) Larvae were scored as without (‘normal’, black) or with loss of eye pigment (grey). (j) Sanger sequencing on individually scored larvae was analyzed by EditR and plotted according to phenotype. Scale bars = 400 µm (c) or 100 µm (i). dpf / hpf = days/hours post fertilization.
|
( |
( |
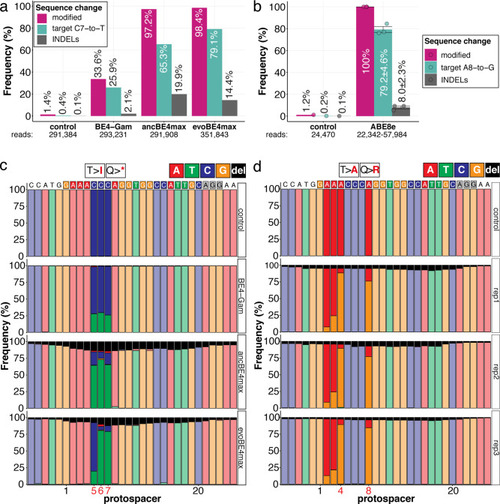
Genomic DNA samples from oca2-Q333 base editing experiments (Figure 2b–g) were used to query the outcome of intended base editing and indel formation by quantitative means by Illumina sequencing of the target region. Note: for oca2-Q333 control, BE4-Gam, ancBE4max, and evoBE4max, two pools of five embryos were used as sample input (a, c), whereas all three biological replicates of ABE8e samples were sequenced separately (b, d). The proportion of all reads aligned per sample to a reference is plotted, distinguishing (1) all modified reads, (2) target cytosine (C7, a) or adenine (A8, b) nucleotide changes and (3) INDELs. The number of reads shown (a, b) refers to all aligned Illumina reads per sample. The frequencies of base calls at the oca2-Q333 sgRNA target site ± 5 bp is shown for the three different cytosine editors (c) and ABE8e (d).
|
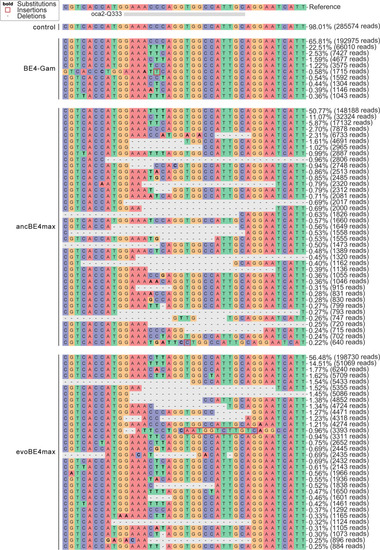
Allele frequency of CBE experiments at the |
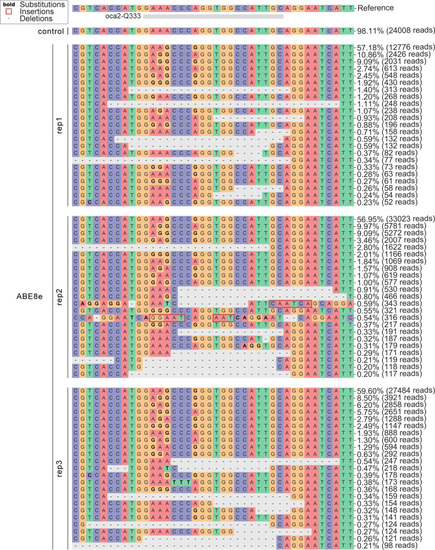
Allele frequency of ABE8e experiments at the |
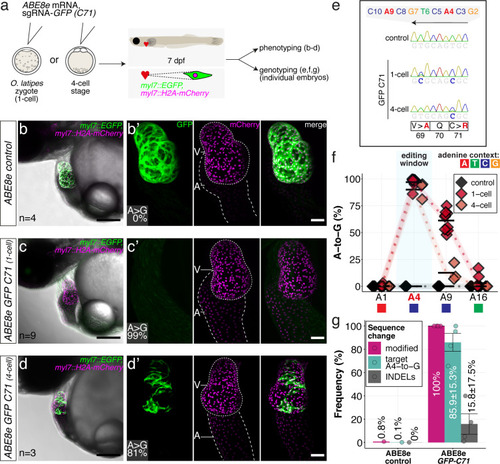
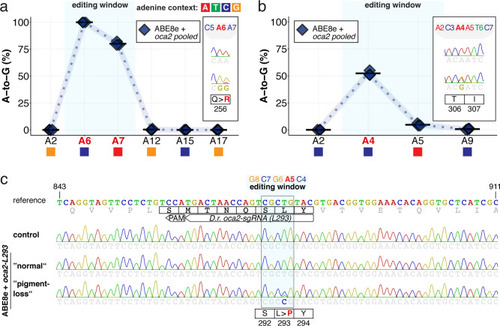
(a) Co-injection of ABE8e mRNA with GFP-C71 sgRNA into a single cell of one-cell or four-cell stage medaka embryos (myl7::EGFP, myl7::H2A-mCherry). Control injections with ABE8e mRNA. Scoring of fluorescence was performed at 7 dpf followed by genotyping of each individual embryo. Confocal microscopy of chemically arrested hearts (representative images) at 7 dpf (lateral view with V = ventricle, A = atrium). Overview images, overlaid with transmitted light, show maximum z-projections of optical slices acquired with a z-step size of 5 µm. Scale bar = 200 µm (b–d). Close-up images show maximum z-projections of optical slices acquired with a z-step size of 1 µm. Note the display of A-to-G conversion rates for A4 causing the p.C71R missense mutation (see g) and Supplementary file 2. Scale bar = 50 µm (b-d’). (e–f) Quantification of Sanger sequencing reads show close to homozygosity rates of A-to-G transversions installing the C71R missense mutation (Supplementary file 2). Note: sgRNA GFP-C71 targets the complementary strand (arrow in f). To highlight the dinucleotide context, the nucleotide preceding the target A is shown by red (A), green (T), blue (C) and yellow (G) squares below the respective A. dpf = days post fertilization. (g) Amplicon-seq of the target region a subset (n = 4) of 1 cell stage ABE8e experiment gDNA samples (single embryos) was used to quantify the outcome of intended base editing and indel formation. Aligned Illumina-reads analyzed, 14,653 (control); 23,201 (ABE8e rep1); 10,696 (ABE8e rep2); 66,311 (ABE8e rep3); 48,126 (ABE8e rep4).
|
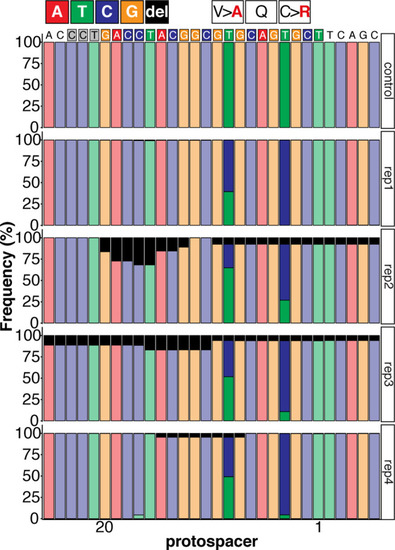
Sequence composition following Amplicon-seq of ABE8e GFP-C71 editants surrounding the GFP-C71 sgRNA target site ±5 bp. |
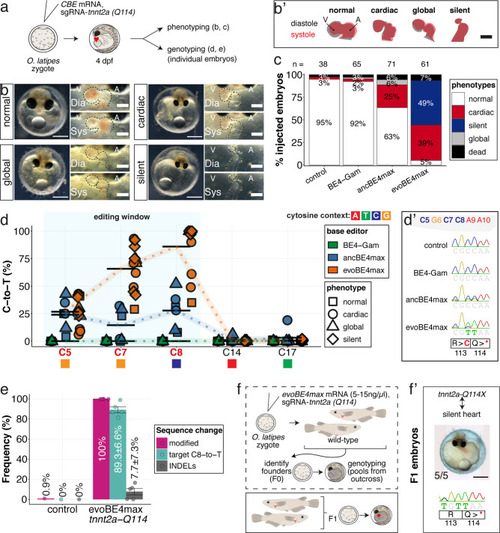
(a) Schematic diagram of protocol after co-injection of cytosine editor mRNA (comparing BE4-Gam, ancBE4max, evoBE4max) with sgRNA tnnt2a-Q114 (PTC) into one-cell stage medaka embryos; control injections only contained the sgRNA. (b) Editing of tnnt2a resulted in a range of phenotypes classified into five categories, including general morphogenic (global), dysmorphic but still functional heart chambers (cardiac), and non-contractile hearts (silent), where cardiac and silent phenotype groups displayed homogeneously additional developmental retardation. Scale bar = 400 µm (overview) and 100 µm (zoom-in). Ventricle (V), atrium (A), diastole (Dia) and systole (Sys) are indicated. (b') Representative scheme of fractional shortening of the heart chambers in specified phenotype groups highlighting significant morphological consequences (small ventricle) in the silent heart group. (c) Fraction of phenotype scores as a consequence of cytosine base editor injections. (d) Summary of editor type-specific C-to-T conversion efficiencies relative to the target C protospacer position grouped by phenotype class for BE4-Gam (n = 6), ancBE4max (n = 6) and evoBE4max (n = 12). To highlight the dinucleotide context, the nucleotide preceding the target C is shown by red (A), green (T), blue (C), and yellow (G) squares below the respective C. (d') Example Sanger sequencing reads of single edited embryos with resulting missense and stop-gain mutations through editing at C5, C7, and C8, respectively (e) Amplicon-seq of the target region of a subset (n = 5) of evoBE4max edited gDNA samples (single embryos) quantified target C8-to-T editing as well as indel frequencies. Aligned Illumina-reads analyzed, 7094 (control); 11,557 (evoBE4max rep1); 2561 (evoBE4max rep2); 2481 (evoBE4max rep3); 37,751 (evoBE4max rep4); 48,791 (evoBE4max rep5). (f) Phenotypic analysis of F1 tnnt2a-Q114X mutants revealed complete penetrance (n = 5) of the silent heart phenotype with same phenotypic profile as for F0 edits. dpf = days post fertilization.
|
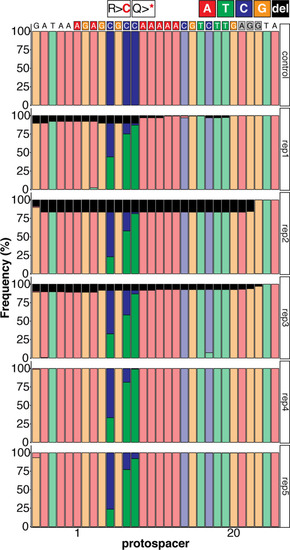
Sequence composition determined by Amplicon-seq of evoBE4max tnnt2a-Q114 editants surrounding the tnnt2a-Q114 sgRNA target site ± 5 bp. |
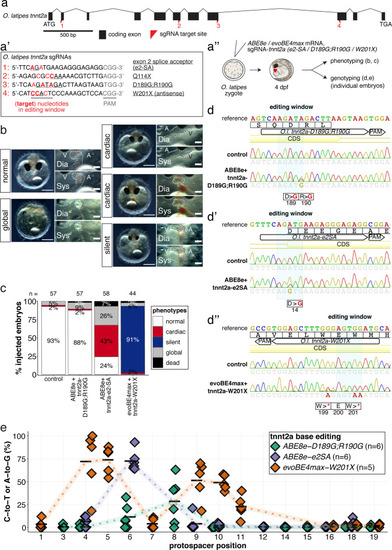
(a) O. latipes tnnt2a coding region with annotation of all sgRNAs (a’) targeting tnnt2a used in this study. (b) Overview of phenotypic categories and the quantification of phenotypic outcomes following base editing F0 tnnt2a base editing experiments (c). (d) Representative Sanger sequencing reads. (e) Summary of C-to-T or A-to-G conversion efficiencies.
|
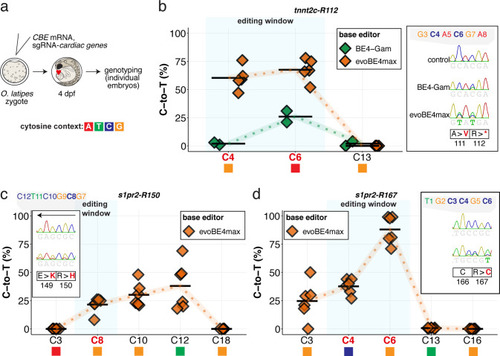
Summary of C-to-T conversion efficiencies. Sample number used for evoBE4max: n = 5, 6, 6, respectively for (b–d); and BE4-Gam: n = 2 (b). To highlight the dinucleotide context, the nucleotide preceding the target C is shown by red (A), green (T), blue (C) and yellow (G) squares below the respective C.
|
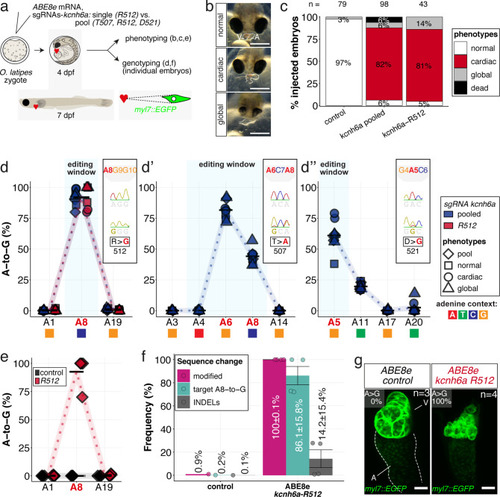
(a) Regime of ABE8e mRNA injections with a single (kcnh6a-R512) or pooled sgRNAs (kcnh6a-T507, -R512, -D521) targeting different amino acid codons in the voltage sensor S4 domain/S4-S5 linker of the medaka potassium channel ERG in myl7::EGFP (Cab strain) transgenic embryos; control injection included ABE8e mRNA only. (b) Phenotypes in F0 comprised primary cardiac malformation (dysmorphic ventricle with impaired contractility) and more severe global phenotypes with general retarded development and prominently dysmorphic hearts the proportions of which are given in (c). Scale bar = 400 µm. (d-d'') Genotyping summaries of the three sgRNA loci with phenotype class annotations for each genotyped specimen with a comparison of single sgRNA-R512 injection to a pool with two additional sgRNAs (T507 and D521) targeting the medaka ERG S4 voltage sensor; inlets display Sanger reads with the editing of A8 (d), A6 and A8 (d') and A5 (d'') contained in the core editing windows; sgRNA pool (n = 8) and sgRNA-R512 (n = 6). To highlight the dinucleotide context, the nucleotide preceding the target A is shown by red (A), green (T), blue (C) and yellow (G) squares below the respective A. (e–g) Confocal microscopy of the heart in a myl7::EGFP reporter line injected with ABE8e mRNA and sgRNA-R512 at 7 dpf reveals significant chamber wall defects of non-contractile/spastic ventricles with A-to-G editing of 100% in 3/4 of the specimen as determined by Sanger sequencing (e). (f) Amplicon-seq of the same gDNA samples (single embryos, n = 4) quantified target A8-to-G editing and indel frequencies. Aligned Illumina-reads analyzed, 11,387 (control); 24,936 (ABE8e rep1); 4038 (ABE8e rep2); 75,148 (ABE8e rep3); 86,327 (ABE8e rep4). Images show maximum z-projections of optical slices acquired with a z-step size of 1 µm (g). Note the display of A-to-G conversion rates. Scale bar = 50 µm. V = ventricle, A = atrium, dpf = days post fertilization.
|
( |
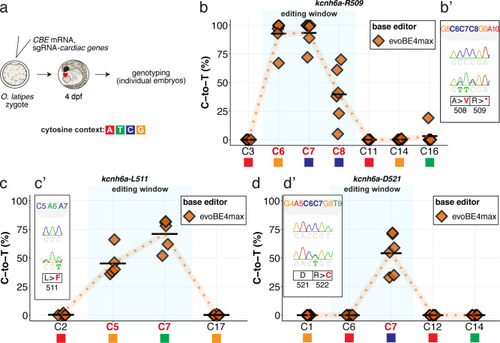
Summary of C-to-T conversion efficiencies relative to the target C protospacer position for Sample number: n = 6, 4, 6 respectively ( |
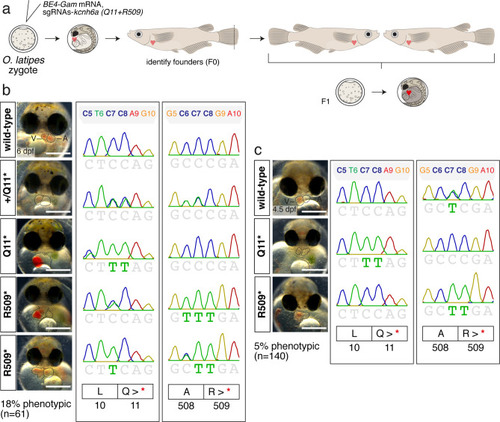
( |
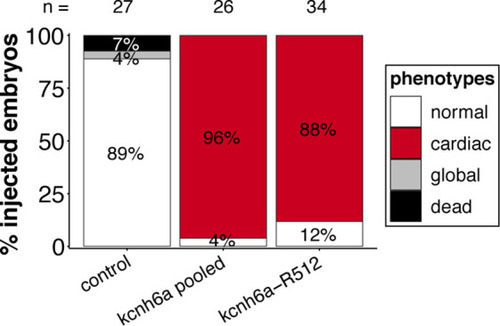
Targeting the three kcnh6a loci simultaneously or kcnh6a-R512 alone in a different genetic background (myl7::EGFP, myl7::H2A-mCherry; HdrR strain) recapitulates phenotypic proportions. |
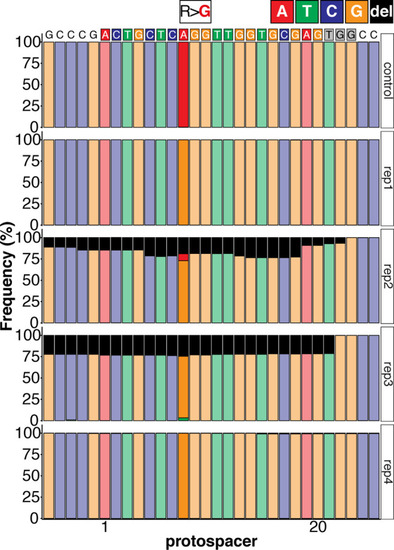
Sequence composition determined by Amplicon-seq of ABE8e kcnh6a-R512 editants surrounding the kcnh6a-R512 sgRNA target site ± 5 bp. |
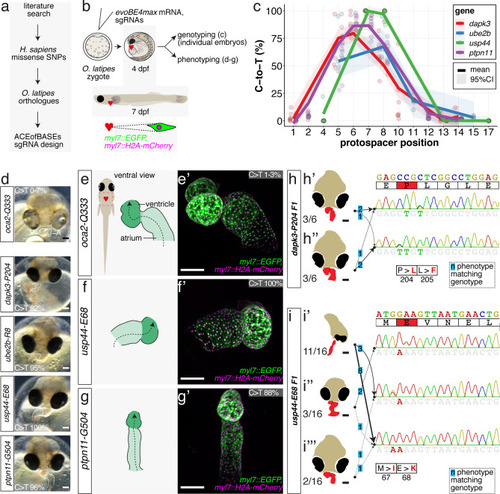
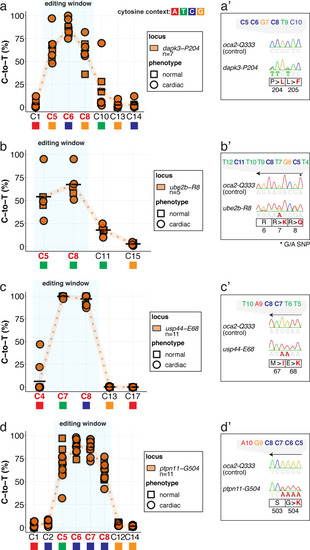
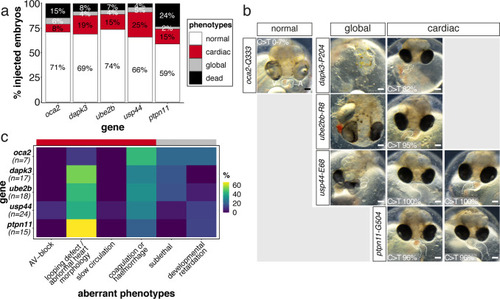
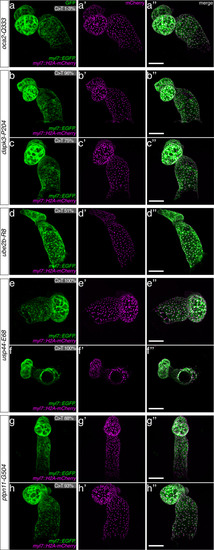
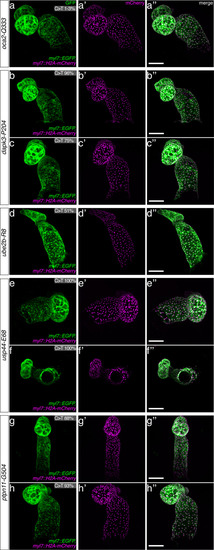
(a) Candidate human CVD gene SNV validation workflow. (b) To target the SNVs evoBE4max mRNA was co-injected into the 1 cell stage of the medaka wild-type or myl7::EGFP, myl7::H2A-mCherry reporter strain together with the corresponding target or oca2-Q333 (control) sgRNAs. Individual, imaged, embryos were then further analyzed to determine the rate of C-to-T transversions. (c) Cytosine editing efficiencies are substantial for all candidate genes tested. Data shown in Figure 7—figure supplement 2 was replotted, including all data points from a-d across all target cytosine along the protospacer. Sample numbers: dapk3-P204 (n = 7), ube2b-R8 (n = 5), usp44-E68 (n = 11), and ptpn11-G504 (n = 11). (d) Representative phenotypes of 4 dpf base edited embryos are shown for all four tested candidate CVD genes including oca2-Q333 controls. Top, ventral view, with V = ventricle, A = atrium. (e–g) Confocal microscopy of selected candidate validations in the reporter background. Hearts were imaged in 7 dpf hatched double fluorescent embryos. Images show maximum projections of the entire detectable cardiac volume with a step size of 1 µm. Cartoons (left) highlight the looping defects observed in usp44 and ptpn11 base edited embryos with ventricle-atrium inversion (f) or tubular heart (g). (e’-g’) Imaged embryos were subsequently genotyped and quantified C-to-T transversions for the target codon are shown. Note: due to the inverted nature of the confocal microscope used, raw images display a mirroring of observed structures, which we corrected here for simpler appreciation. Phenotypic analysis of F1 dapk3-P204L (h) and usp44-E68K (i) embryos revealed that homozygous changes at P204L or E68K lead to cardiac malformations with varying degree: looping (h’) and mild looping defects (h’’, i’’’); altered heart morphology (i’’). Bystander edits (hetero- or homozygous, usp44-E68K) lead to additional developmental defects, including brain and eye abnormalities (i’). Scale bar = 100 µm (d, e–i). dpf = days post fertilization.
|
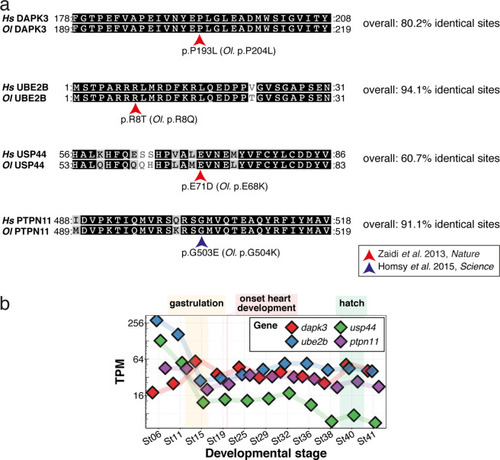
( |
( |
( |
( |
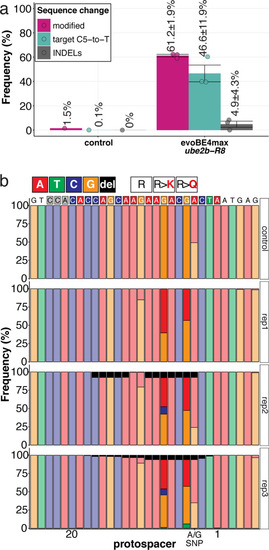
The full complement of analyzed CVD associated phenotypes in cytosine base editing of |
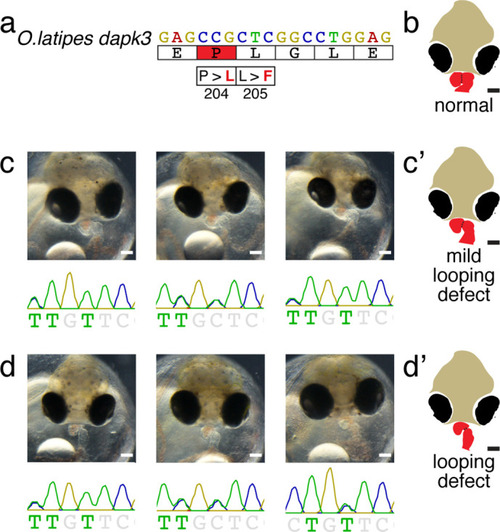
Cartoons ( |
Cartoons (c’, d’ and e’) are shown in the main figure as summary of the phenotypic manifestation. |
( |