
- Title
-
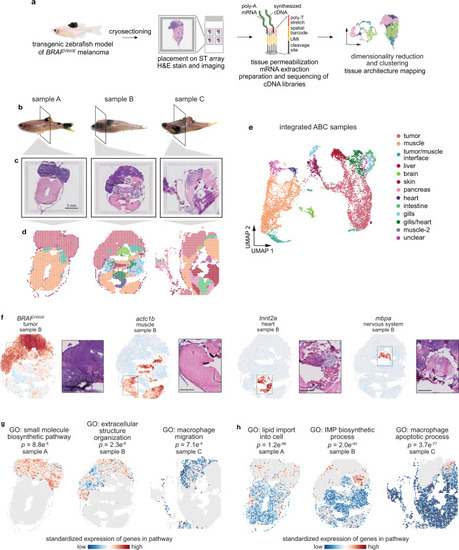
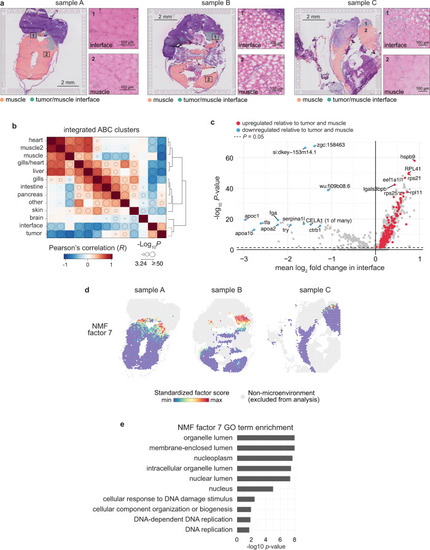
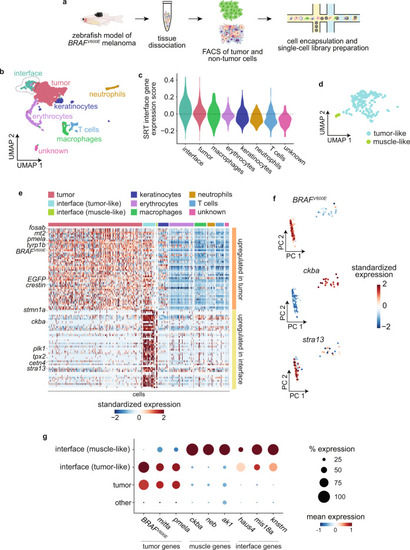
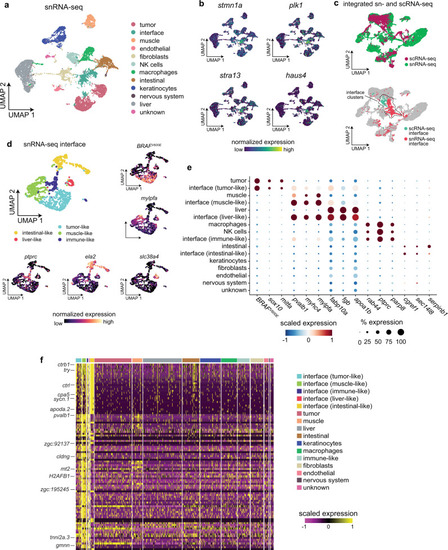
Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface
- Authors
- Hunter, M.V., Moncada, R., Weiss, J.M., Yanai, I., White, R.M.
- Source
- Full text @ Nat. Commun.
|
PHENOTYPE:
|
|
|
|
PHENOTYPE:
|