
- Title
-
Selective and competitive functions of the AAR and UPR pathways in stress-induced angiogenesis
- Authors
- Zhang, F., Zeng, Q.Y., Xu, H., Xu, A.N., Liu, D.J., Li, N.Z., Chen, Y., Jin, Y., Xu, C.H., Feng, C.Z., Zhang, Y.L., Liu, D., Liu, N., Xie, Y.Y., Yu, S.H., Yuan, H., Xue, K., Shi, J.Y., Liu, T.X., Xu, P.F., Zhao, W.L., Zhou, Y., Wang, L., Huang, Q.H., Chen, Z., Chen, S.J., Zhou, X.L., Sun, X.J.
- Source
- Full text @ Cell Discov
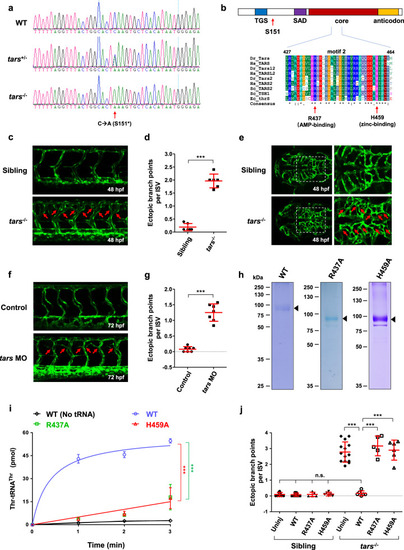
Angiogenic phenotypes of the tars mutants and the requirement of Tars aminoacylation activity.
a Sequencing results of the wild-type (WT) and the tars mutated heterozygous (tars+/−) and homozygous (tars−/−) zebrafish embryos. The arrow denotes the C-to-A nonsense mutation that generates a premature stop codon after serine 151 (S151*). b Domain architecture of the Tars protein, showing the position of serine 151 (S151; arrow), where the mutation-introduced stop codon predicts a deletion of the downstream SAD, core, and anticodon domains. Also shown is the alignment of the amino acid sequences of a signature motif (motif 2) within the core domain. Two evolutionarily conserved residues, R437 and H459, which are required for the aminoacylation activity and are directly involved in AMP and zinc binding, respectively, were chosen for functional studies. The alignment includes Tars and its homologs (i.e., Tars2 and Tarsl2, etc.) from humans (Hs, Homo sapiens), zebrafish (Dr, Danio rerio), yeast (Sc, Saccharomyces cerevisiae), and enterobacterial (Ec, Escherichia coli). c Confocal microscopy images of EGFP-labeled blood vessels in the trunk of the WT and tars−/− zebrafish embryos at 48 hpf. d Quantification and statistical analysis of the ectopic branch points per ISV of the tars−/− and sibling embryos. e An abnormal increase of branches in the hindbrain capillaries of the tars−/− embryos compared with the siblings. Magnified views of the dashed boxed regions are shown on the right. f Increased branch points in the ISVs caused by tars MO in WT embryos. In panels c, e, f, the arrows denote ectopic branch points of the vessels. g Quantification and statistical analysis of the ectopic branch points per ISV of the control and Tars knockdown embryos. h Coomassie blue staining of WT and mutant zebrafish Tars proteins, which were purified with His-tag from E. coli. i Aminoacylation activity assays with the purified proteins, showing the nearly abolished enzymatic activities of the R437A and H459A mutants compared with the WT Tars protein. j Rescue of the tars−/− angiogenic phenotype by injection of the WT, but not the inactivation mutant, tars mRNAs. Note that, for the tars−/− embryos, injection of the WT tars mRNA, but not the R437A or H459A mutant mRNA, significantly reduced the ectopic branch points per ISV compared with the uninjected tars−/− embryos (uninj). In contrast, for the WT embryos, injection of these WT, R437A, and H459A mRNAs showed no effect on the ISVs compared to the uninjected controls. Also, note that the phenotypic rescue by the WT tars mRNA was almost complete because quantification of the branch points of the injected embryos showed no difference compared with the WT embryos. In panels d, g, i, j, data are presented as means ± SD; two-tailed t-test; ***P < 0.001; n.s., not significant. |
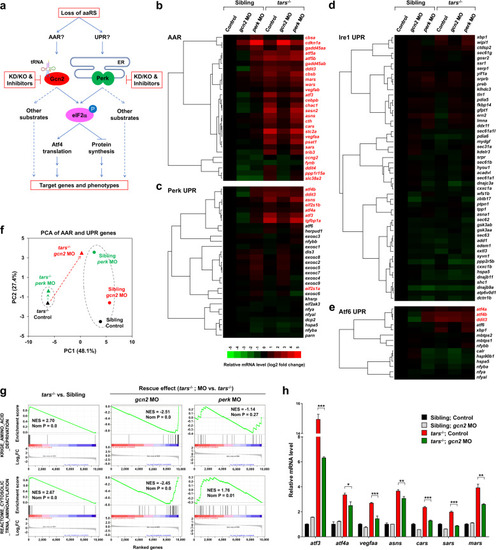
Differential and combinatorial regulation of the AAR- and UPR-associated genes by functional loss of Tars, Gcn2, or Perk.
a Schematic overview of study design. Both AAR and UPR may regulate the angiogenic phenotypes induced by deficiency of aminoacyl-tRNA synthetases (aaRSs). To distinguish between them, the proposed experimental strategies (in red boxes) were based on the tars mutant and the siblings; knockdown (KD), knockout (KO), and pharmacological inhibition of Gcn2 and Perk were used, and systematic gene expression profiling of the zebrafish embryos with various genotypes and treatments was performed. b–e Hierarchical clusters and heatmaps of the expression levels of the genes in the AAR gene set (b) and in the Perk-, Ire1-, and Atf6-mediated UPR gene sets (c–e). The data were produced by RNA-seq analyses of 36 hpf homozygous tars-mutated (tars−/−) embryos and their siblings from the same litters (sibling) that were treated with indicated morpholino (MO). The gene symbols written in red are known to be involved in AAR, even though some of them are also listed in the UPR gene sets (for further information and references, see Supplementary Tables S1-S4). The color bar indicates relative expression levels. Note that the upregulated genes in tars−/− mostly fall into the AAR category (written in red), whereas the UPR genes are largely unchanged, except for those shared in both pathways. f PCA of the AAR- and UPR-associated genes of the tars−/− (triangles) and siblings (circles) treated with gcn2 MO (red) or perk MO (green), compared with the control groups (black), respectively. Note that gcn2 MO, but not perk MO, significantly reversed the major component (PC1) score of tars−/−, whereas the perk MO only altered the PC2 score of the siblings but has a very subtle effect on the tars-mutants. g GSEA results showing strong enrichments of the AAR and tRNA aminoacylation genes in the tars−/− embryos, which can be reversed by gcn2 MO but not perk MO. h RT-qPCR analysis of the representative genes that are activated in the tars−/− embryos and are downregulated by gcn2 MO. Data are presented as means ± SD of triplicate reactions. ***P < 0.001; **P < 0.01; *P < 0.05. The P values for the increased expression of all these genes in tars−/− relative to the siblings are less than 0.001 (not shown). |
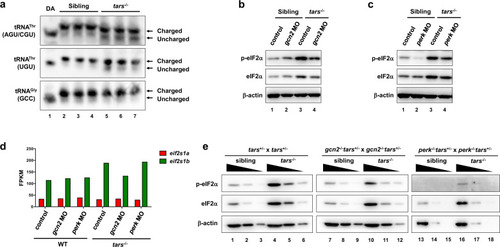
Selective activation of Gcn2 and Perk in stress condition and normal development.
a Northern blot results showing the increased uncharged tRNAThr in the tars−/− embryos compared with the siblings. Charged and uncharged tRNAs (upper and lower bands, respectively) were separated in an acid polyacrylamide/urea gel system; the three types of tRNAThr were hybridized with specific probes that could recognize tRNAThr(AGU/CGU) or tRNAThr(UGU). The tRNAGly(GCC) was used as a negative control. Deacylated tRNAs (DA) were used to mark the migration position of the uncharged tRNAs on the gels. b, c Immunoblot analysis of the phosphorylated eIF2α (p-eIF2α) and total eIF2α upon morpholino-mediated knockdown of Gcn2 or Perk in the WT and tars−/− embryos. β-actin was used as the loading control. Note that gcn2 MO and perk MO reduced the phosphorylated eIF2α in the tars−/− embryos to a comparable extent, whereas only perk MO reduced the eIF2α phosphorylation in the WT embryos. d Expression levels of the mRNAs of the two zebrafish eIF2α-coding genes, eif2s1a and eif2s1b, in the embryos of indicated genotypes. Presented are fragments per kilobase per million mapped reads (FPKM) values from RNA-seq data. Note that eif2s1b is upregulated in the tars−/− embryos, which explains the accordingly increased protein levels as indicated by the immunoblot results. e Immunoblot analysis of the phosphorylated and total eIF2α level in the gene knockout embryos, which were produced by crossing the indicated mutant lines. Each sample was loaded in a 3-fold serial dilution to facilitate quantification. Note that the basal level of p-eIF2α was hardly detectable in the perk−/− “normal” (sibling) embryos (lanes 13–15), and that the p-eIF2α in the gcn2−/−tars−/− embryos was decreased, but not eliminated, compared with the tars−/− embryos (compare lanes 7–12 with 1–6). |
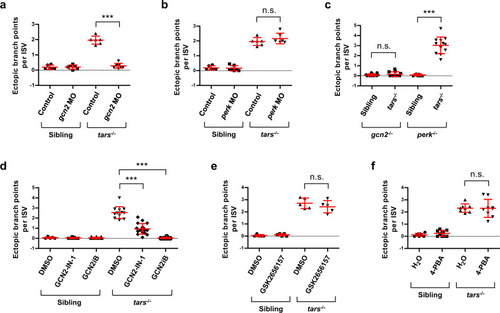
Genetic and pharmacological inhibition of AAR, but not UPR, suppresses the tars-deficiency-induced angiogenesis.
a, b Quantification and statistical analysis of the ectopic branch points per ISV of the tars−/− and sibling embryos that were treated with gcn2 MO or perk MO. c Knockout of gcn2, but not perk, suppressed the tars-deficiency-induced angiogenesis. The embryos were produced by self-cross of gcn2−/−tars+/− or perk−/−tars+/− lines, and the tars−/− ones were compared with their siblings in the same litter. d Pharmacological inhibition of Gcn2 by GCN2-IN-1 and GCN2iB significantly suppressed the angiogenic phenotypes of the tars−/− embryos. e Inhibition of Perk by Perk-specific inhibitor GSK2656157 showed no significant rescue effect on the angiogenetic phenotypes. f Attenuation of UPR by 4-phenylbutyrate (4-PBA) could not rescue the angiogenetic phenotypes. In panels d–f, the solvents DMSO and H2O were applied as negative controls, respectively. In the statistical analyses, data are presented as means ± SD; two-tailed t-test; ***P < 0.001; n.s., not significant. PHENOTYPE:
|
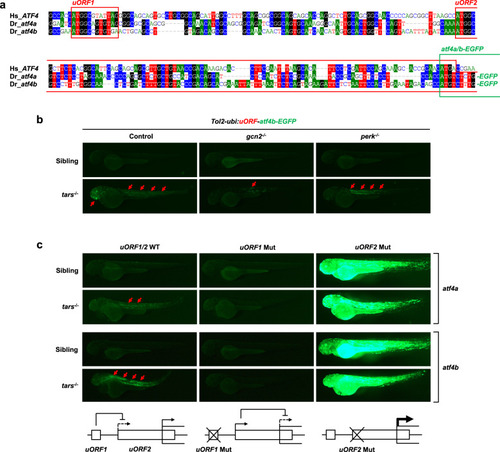
The tars-deficiency-induced angiogenesis is dependent on the ribosome re-initiation mechanism of Atf4 translational regulation.
a An alignment of the sequences of the translational regulatory regions of human ATF4 and zebrafish atf4a and atf4b, showing the highly conserved upstream open reading frames (uORF1 and uORF2; red boxes) and coding ORFs (green box). These regions of zebrafish atf4a and atf4b were cloned into the reporter vectors, and their coding ORFs were fused with the ORF of EGFP. b Fluorescence microscopy images showing the enhanced expression (i.e., translation) levels of the atf4a- and atf4b-EGFP fusion ORFs (red arrows) in the truck and head of tars−/− embryos, which were inhibited by the knockout of gcn2, but not perk. The Tol2 transposase-based transgenic system containing a ubiquitin promoter (ubi:) was used to drive the expression of the reporters. c Mutation analysis of the zebrafish atf4a and atf4b uORFs for their eIF2α phosphorylation-dependent translational regulation. The start codon (ATG) of uORF1 or uORF2 was mutated into AGG. The expression levels of the WT and uORF1- and uORF2-mutated (Mut) atf4a- and atf4b-EGFP reporters were demonstrated by representative fluorescence microscopy images. The lower panels present the working model in which uORF1 can reduce the usage of the inhibitory uORF2, so that the uORF1 Mut eliminates the AAR-dependent upregulation of atf4a/b expression, whereas the uORF2 Mut leads to an extremely high AAR-independent atf4a/b expression. PHENOTYPE:
|
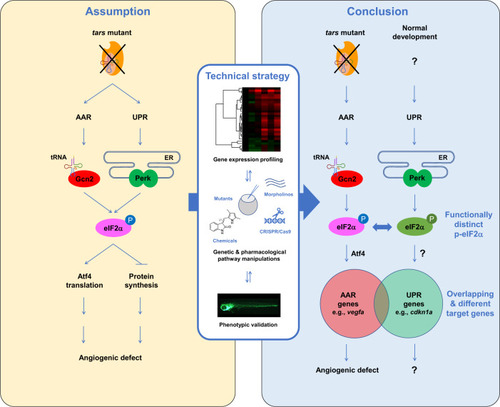
A summary of the study and the working model for the selective and competitive functions of AAR and UPR pathways.
The close interconnection of the AAR and UPR pathways makes it difficult to distinguish between them, therefore the angiogenesis induced by aaRS deficiency has been assumptively attributed to activation of both pathways. While UPR has been addressed in previous studies, it is unclear whether AAR is also activated and whether both are required in this process (left panel). We established that these highly interconnected pathways can be distinguished in the herein-generated zebrafish angiogenic model that harbors a tars mutation, by using an approach combining systematic gene expression profiling and quantitative phenotypic analysis upon a variety of genetic and pharmacological manipulations of these pathways (middle panel). We found that AAR, but not UPR, is activated and is functionally required for the angiogenic phenotypes in the tars mutants (right panel). Notably, while Perk-mediated UPR is inactive in the tars mutants, it plays an important role in normal development; however, the function of Perk is overwhelmed by Gcn2 in the stress condition, through competing for phosphorylation of their shared target, eIF2α. The phosphorylated eIF2α (p-eIF2α) by Gcn2 and Perk can be distinguished by the cells/organisms (therefore illustrated in different colors) and thus regulate the partially overlapped AAR- and UPR-associated genes. The question marks denote the possible cause, mechanism, and functional consequence of UPR, which should be different from those of AAR as addressed in this study. |