
- Title
-
Zebrafish Cancer Predisposition Models
- Authors
- Kobar, K., Collett, K., Prykhozhij, S.V., Berman, J.N.
- Source
- Full text @ Front Cell Dev Biol
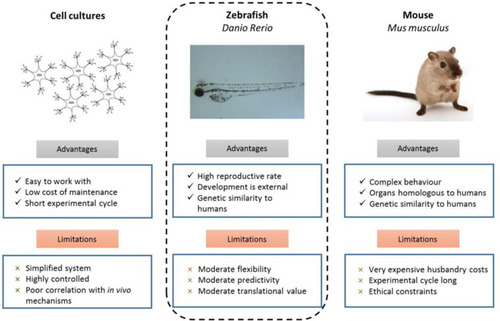
Experimental advantages and limitations of commonly used model organisms ( |
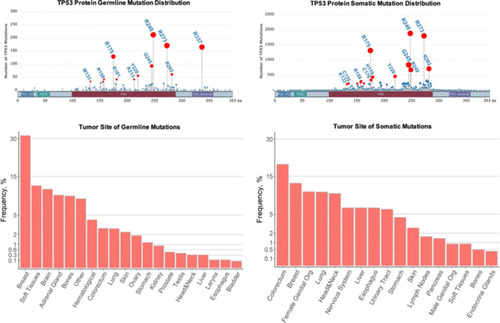
Comparison of germline and sporadic |
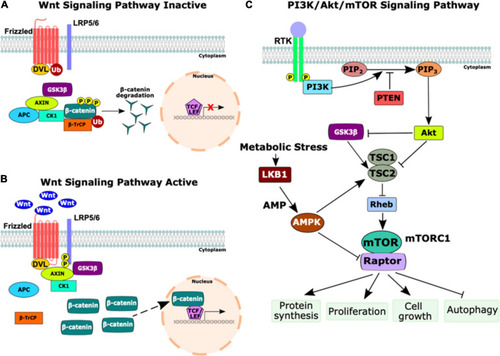
Schematic of the Wnt and PI3K/Akt/mTOR signaling pathways. |
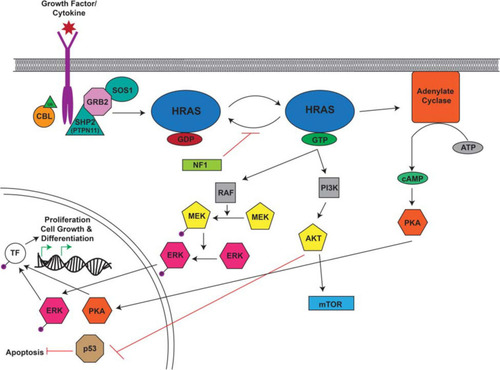
Overview of the RAS Signaling Pathway. The Ras signaling cascade is stimulated by the binding of a growth factor/cytokine to the appropriate RTK. C-CBL (Casitas B-lineage lymphoma) is an E3 ubiquitin ligase responsible for the ubiquitination of RTKs leading to their internalization and degradation. The activated receptor binds the adaptor proteins GRB2 (growth factor receptor bound protein 2) and SHP2/PTPN11 (protein tyrosine phosphatase non-receptor type 11) and recruits SOS1 (son of sevenless homolog 1). SOS1 facilitates the conversion of RAS from its inactive to active confirmation through the binding of GTP. RAS-GTP activates RAF, PI3K, and adenylate cyclase to cause a signaling cascade. RAF activates MEK which in turn activates ERK. Adenylate cyclase facilitates the production of cAMP which ultimately activates PKA. Activated ERK and PKA are protein kinases that translocate to the nucleus where they phosphorylate and activate transcription factors (TF) which drive the transcription of target genes involved in proliferation, cell growth, and differentiation. PI3K activates AKT which can activate mTOR and inhibit p53, leading to the inhibition of apoptosis. This figure was generated using information from several research articles and reviews ( |
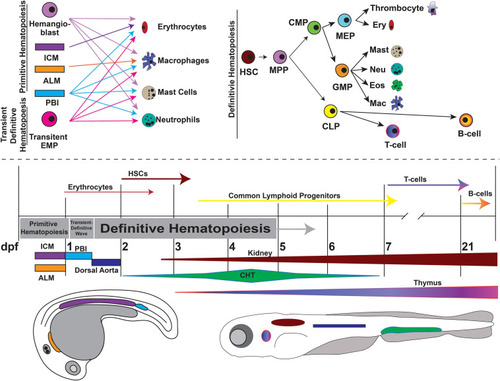
Overview of zebrafish hematopoiesis. Zebrafish hematopoiesis occurs in two waves, primitive and definitive, the latter of which recapitulates all the same blood cell types as humans. During the primitive wave, hemangioblasts give rise to a portion of primitive erythrocytes and granulocytes. The main source of primitive granulocyte production occurs within the anterior lateral mesoderm (ALM) (orange), while primitive erythrocyte productions occurs in the intermediate cell mass (ICM) (purple). Once circulation begins at approximately 24 hpf, primitive blood production transitions to the posterior blood island (PBI) (blue). Between 24 and 36 hpf a transient wave occurs in which transient bipotential erythromyeloid progenitor cells (EMPs). By 36 hpf, the first hematopoietic stem cells (HSCs) arise from the ventral wall of the dorsal aorta (navy), a site analogous to the aorta-gonad-mesonephros (AGM) in mammals. Once HSCs arise, they migrate to the caudal hematopoietic tissue (CHT) (previously the PBI), a structure analogous to the mammalian fetal liver. It is in the CHT that definitive erythroid and myeloid cells are produced and replace their primitive counterparts. By 4dpf HSCs migrate to and seed the kidney marrow (dark red), the equivalent of bone marrow in mammals. The first T-cells originate at approximately 7 dpf and mature in the thymus (purple/pink), while B-cells are not present until 21 dpf. This figure was constructed using information from published reviews ( |
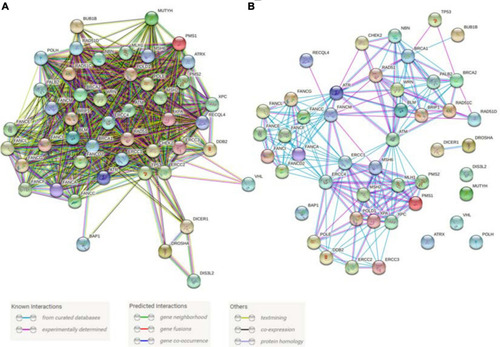
Predicted and known interactions among the cancer predisposition genes involved in DNA repair and genome stability maintenance. |
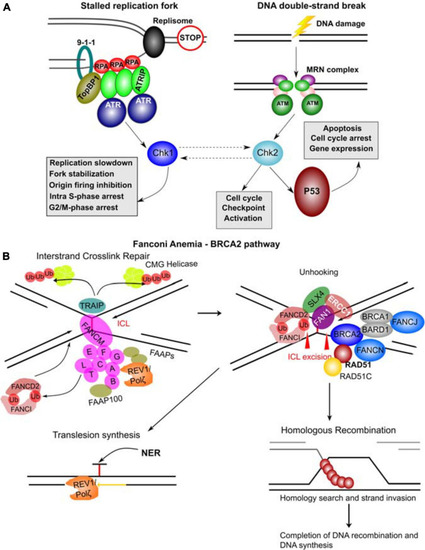
Molecular complexes formed and DNA repair pathways mediated by CPG proteins. (A) Mechanisms of ATR and ATM activation by different types of DNA damage. Stalled replication forks resulting from a blockage of replisome progression (indicated by the “STOP” diagram). The single-stranded DNA (ssDNA) region is recognized by Replication Protein A (RPA) forming the filaments which can then recruit ATR-ATRIP (ATR Interacting Protein) complexes. The RAD17-RFC complex (not shown) loads the 9-1-1 complex (RAD9-RAD1-HUS1) onto the ssDNA-dsDNA junction. RAD9 S387 phosphorylation provides a binding site for DNA topoisomerase 2-binding protein 1 (TopBP1), which in turn binds and activates ATR kinase. ATR phosphorylates and activates Chk1 kinase mediating many of its functional effects and directly phosphorylates many other target proteins. DNA double-strand breaks results in a complex signaling cascade resulting in binding of the MRN complex (MRE11-RAD50-NBN) to the DNA ends. MRN provides a DNA damage sensor platform for recruitment and activation of ATM, which then phosphorylates multiple targets including Chk2 responsible for cell cycle checkpoint activation and p53 activation. Chk2 and Chk1 can also functionally interact (hashed lines with arrows). p53 activation leads to apoptosis, cell cycle arrest and gene expression changes. (B) FA – BRCA2 pathway. A major role of this pathway is to respond to inter-strand crosslinks (ICL), which can be especially problematic during DNA replication since ICLs prevent replication fork convergence. At such blocked replication forks, TRAIP ubiquitin ligase ubiquitinates the CMG (CDC45-MCM-GINS) Helicase leading to its unloading. The ICL itself is recognized by FANCM / FAAP24 complex. FANCM then recruits the core FA complex components and FAAPs (FA associated proteins). E2 ubiquitin-conjugating enzyme FANCT and E3 ubiquitin ligase FANCL then cooperate to perform mono-ubiquitination of FANCD2 and FANCI forming the ID2 complex. Monoubiquitinated ID2 complex gets recruited the ICL lesion. In the process of unhooking, DNA near the ICL gets processed by nucleases to enable specific DNA repair pathways. The strand with the ICL structure gets repaired by Nucleotide Excision Repair (NER) and the translesion synthesis by REV1/Polζ, which gets recruited by the FA core complex. ID2 recruits FANCP (SLX4), FANCQ (ERCC1) and the nuclease FAN1 to mediate the ICL incision. The ID2 also interacts with BRCA2 (FANCD1), which gets recruited to the parts of blocked replication forks that will have to go through DNA recombination. BRCA2 in complex with BRCA1, BARD1, FANCN, FANCJ, RAD51 and RAD51C as well as other factors leads to loading of RAD51 into resected ssDNA regions, which can then participate in homologous recombination (HR). The figure was constructed based on the information in several recent reviews (Awasthi et al., 2016; Fradet-Turcotte et al., 2016; Bonilla et al., 2020; Liu et al., 2020). |