
- Title
-
Glutamate 73 Promotes Anti-arrhythmic Effects of Voltage-Dependent Anion Channel Through Regulation of Mitochondrial Ca2+ Uptake
- Authors
- Shimizu, H., Huber, S., Langenbacher, A.D., Crisman, L., Huang, J., Wang, K., Wilting, F., Gudermann, T., Schredelseker, J., Chen, J.N.
- Source
- Full text @ Front. Physiol.
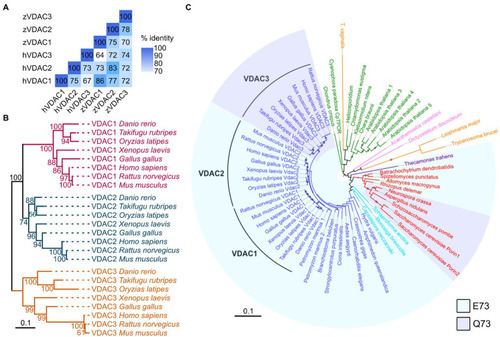
VDACs are highly conserved proteins. |
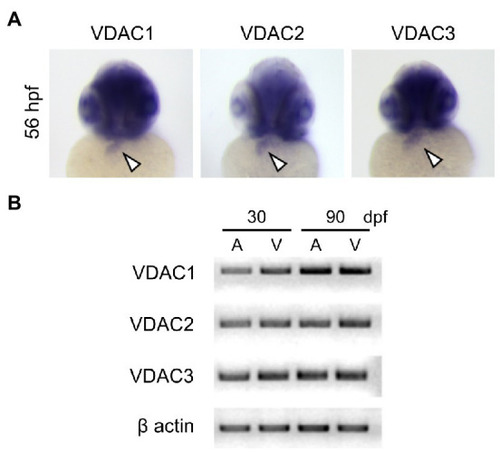
Expression of VDAC isoforms during zebrafish embryonic development. |
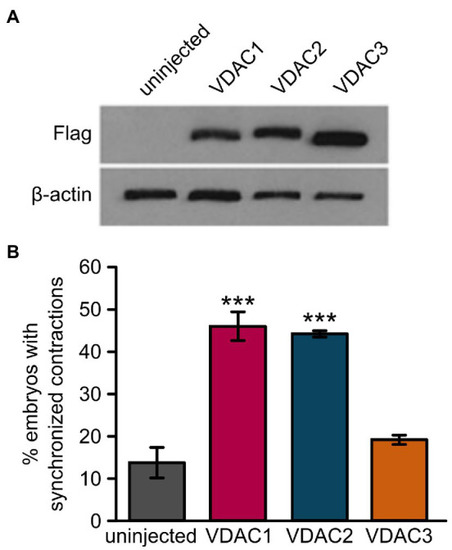
Overexpression of VDAC1 or VDAC2 but not VDAC3 restores rhythmic cardiac contractions in |
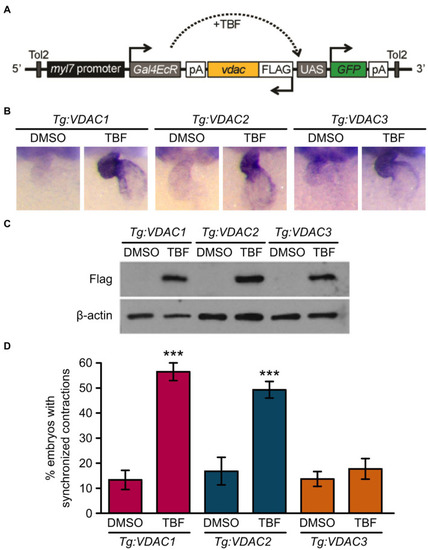
Induction of VDAC1 and VDAC2 but not VDAC3 expression restores rhythmic cardiac contraction in transgenic |
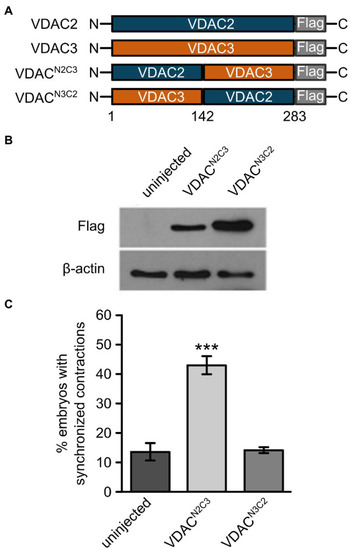
The N-terminal domain of VDAC2 contains critical elements for its cardioprotective activity. |
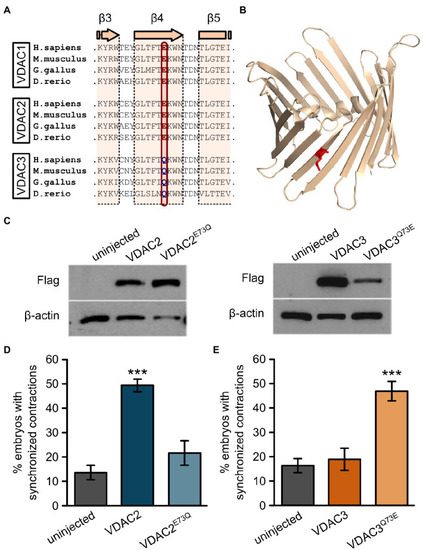
Figure 6. E73 is the critical amino acid residue that determines the ability of VDAC2 to suppress cardiac fibrillation in the tremblor/ncx1h mutant. (A) Alignment of protein sequences of VDAC1, 2, and 3 from different species. In all the species examined the position corresponding to zebrafish residue 73 is consistently occupied by a glutamate (E) in VDAC1 and VDAC2, whereas this position is occupied by glutamine (Q) in VDAC3. (B) Three-dimensional model of the VDAC2 protein (pdb: 4bum) showing the location of amino acid 73 in β-sheet 4 in red. (C) Western blot analysis of lysates from 30 hpf uninjected embryos or embryos injected with 25 pg. FLAG-tagged wild type and point mutant VDAC mRNA. β-actin was used as a loading control. (D) Mutation of E73 to Q in VDAC2 abrogates its ability to suppress cardiac fibrillation in tremblor/ncx1h mutants (49.7 ± 2.8%, N = 3, n = 144 with VDAC2 in contrast to 21.7 ± 5.1%, N = 3, n = 155 with VDAC2E73Q). (E) Vice-versa, by mutating Q73 to E, VDAC3 gained the ability to restore rhythmic cardiac contraction in tre mutants (19.0 ± 4.4%, N = 3, n = 182 with VDAC3 in contrast to 47.2 ± 4.3%, N = 3, n = 145 with VDAC3Q73E). Overall rescue percentages represent the mean rescue percentage ± s.e.m. from N independent experiments, using a total of n embryos. |
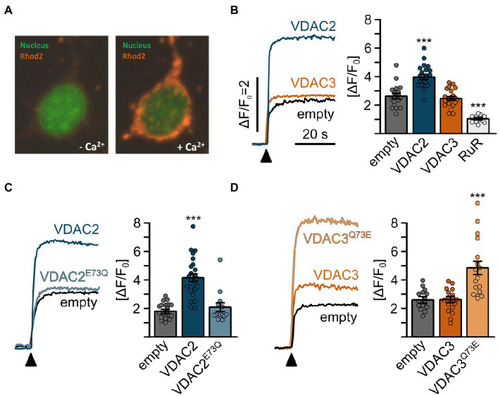
Mitochondrial Ca2+ uptake in HeLa cells is promoted by E73. |
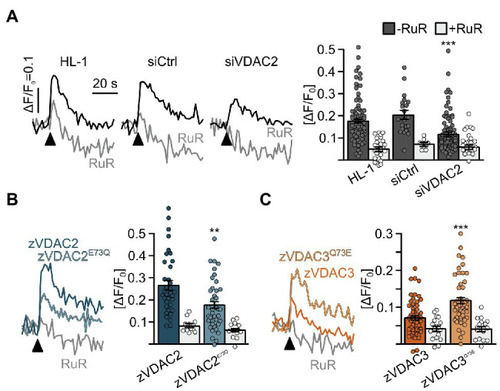
Figure 8. Restoration of SR-mitochondria Ca2+ transfer in permeabilized HL-1 cardiomyocytes. (A) Knockdown of the endogenous mVDAC2 in HL-1 cells significantly reduced SR-mitochondria Ca2+ transfer after addition of caffeine (arrowhead) from ΔF/F0 = 0.18 ± 0.01 (n = 84) to 0.12 ± 0.01 (n = 83) while a scrambled siRNA control did not affect Ca2+ transfer (0.20 ± 0.02, n = 21). (B) Wild-type zVDAC2 restored mitochondrial Ca2+ uptake to 0.27 ± 0.02 (n = 36), well above mVDAC2 knockdown cells. Overexpression of zVDAC2E73Q restored mitochondrial Ca2+ uptake to only 0.18 ± 0.02 (n = 39), which is significantly lower compared to wild-type zVDAC2. (C) Conversely, zVDAC3 did not restore mitochondrial Ca2+ uptake in mVDAC2 knockdown cells (0.12 ± 0.01, n = 54), while VDAC3Q73E restored mitochondrial Ca2+ uptake to ΔF/F0 = 0.20 ± 0.02, (n = 50). Arrowheads indicate injection of 10 mM caffeine. Statistical analysis was performed using Kruskal-Wallis test with Dunn’s post-hoc test. |