
- Title
-
Dynamic changes in the epigenomic landscape regulate human organogenesis and link to developmental disorders
- Authors
- Gerrard, D.T., Berry, A.A., Jennings, R.E., Birket, M.J., Zarrineh, P., Garstang, M.G., Withey, S.L., Short, P., Jiménez-Gancedo, S., Firbas, P.N., Donaldson, I., Sharrocks, A.D., Hanley, K.P., Hurles, M.E., Gomez-Skarmeta, J.L., Bobola, N., Hanley, N.A.
- Source
- Full text @ Nat. Commun.
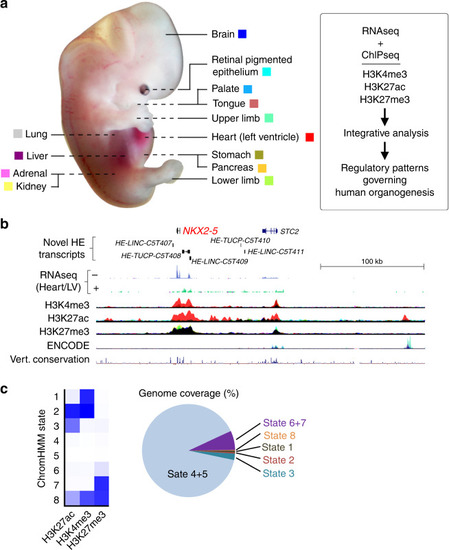
|

|

|

H3K27ac-marked regions were tested in multiple lines of stable transgenic zebrafish (details in Supplementary Data |
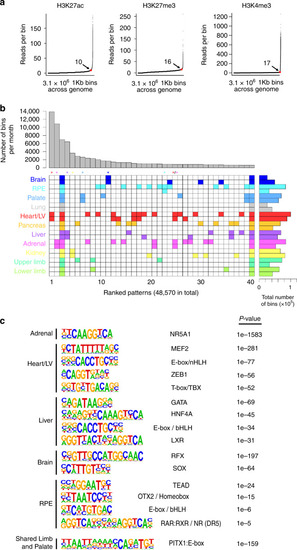
|

|

An intergenic G-to-T de novo mutation (DNM; hg38, chr16:72427838) is shown for a patient with a neurodevelopmental phenotype. Tracks are shown demonstrating additional human embryonic non-coding transcription (enriched in human embryonic brain), the three epigenomic marks, ENCODE data |

|