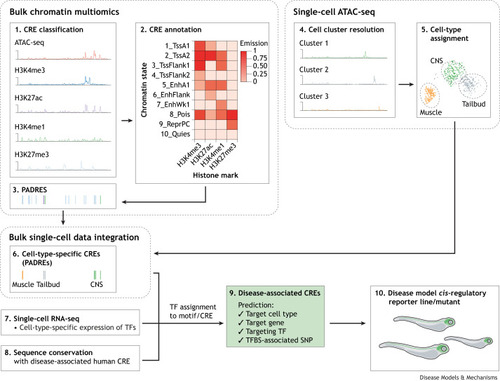
The main features of regulatory elements as determined by DANIO-CODE. (A) Regulatory elements defined by PADREs in DANIO-CODE are identified by ATAC-seq, which assesses chromatin accessibility, and ChIP-seq, which assesses histone modifications, and their function (i.e. level of active transcription) is predicted by the computational tool ChromHMM. This approach has allowed the identification and characterisation of candidate regulatory regions within the zebrafish genome, as shown in the schematic. Further considerations from the DANIO-CODE-available data are also included here. Within the promoter region, transcription of mRNA by RNA polymerase II occurs at the accessible TSS that is defined using CAGE-seq. The example promoters shown, TssA1 and TssA2, are accessible due to histone methylation (H3Kme3 or H3Kme1) and acetylation (H3K27ac), which make the chromatin less condensed. From the active enhancer region, eRNA is bidirectionally transcribed, which is also detected by CAGE-seq. The active enhancers shown, EnhA1 and EnhFlank, have H3Kme1 and H3K27ac histone modifications. Prior to activation, enhancers can exist in a primed state, known as primed enhancers, such as EnhWk1, which is associated with the H3Kme1 histone mark only. Promoter–enhancer interaction spans are detected by chromosome conformation capture techniques, 4C-seq and Hi-C, that can analyse interactions between genomic regions and the three-dimensional architecture of the chromatin. ChIP-seq can also be used to detect CTCFs, which are zinc-finger proteins that bind to highly conserved sequences and act as promoter–enhancer insulators. Finally, transcription can be repressed in polycomb-repressed regions by specific histone marks, such as H3K27me3 for ReprPC, which make the chromatin more condensed. (B) ChromHMM states have been included on the right for further reference (Baranasic et al., 2022). Each of the states are characterized by different levels of histone marks. The left side of this panel shows the different biologically relevant functions assigned to these states. ATAC-seq, assay for transposase-accessible chromatin with sequencing; CAGE-seq, cap analysis of gene expression; ChIP-seq, chromatin immunoprecipitation with sequencing; CTCF, CCCTC-binding factor; EnhA1, active enhancer 1; EnhFlank, enhancer flanking; EnhWk1, weak enhancer; eRNA, enhancer RNA; PADRE, predicted ATAC-seq-supported developmental regulatory element; Pois, poised; Quies, quiescent; ReprPC, repressed polycomb; TSS, transcription start site; TssA1/2, active transcription start site 1/2; TssFlank1/2, TSS flanking.
|