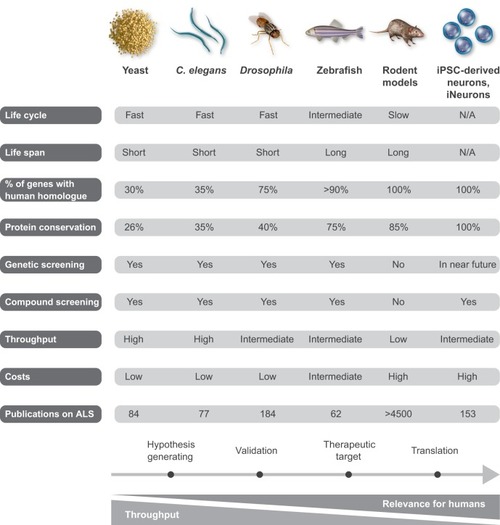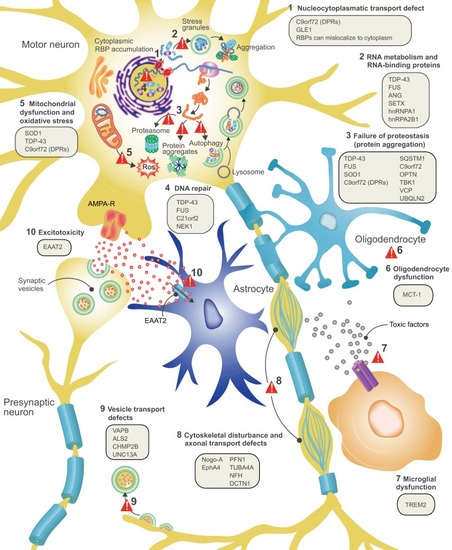
ALS disease pathology and proposed disease mechanisms. At the level of cell pathology, ALS is characterized by axonal retraction and cell body loss of upper and lower motor neurons, surrounded by astrogliosis and microgliosis (see Box 2), with ubiquitin- and p62-positive inclusions in surviving neurons. Proposed disease mechanisms contributing to motor neuron degeneration are: (1) Alterations in nucleocytoplasmic transport of RNA molecules and RNA-binding proteins. (2) Altered RNA metabolism: several important RNA-binding proteins become mislocalized in ALS, with cytosolic accumulation and nuclear depletion. The nuclear depletion causes defects in transcription and splicing. Some RNA-binding proteins can undergo liquid-liquid phase separation and can be recruited to stress granules (TDP-43, FUS, ATXN2, hnRNPA1/A2). Altered dynamics of stress granule formation or disassembly can propagate cytoplasmic aggregate formation. (3) Impaired proteostasis with accumulation of aggregating proteins (TDP-43, FUS, SOD1, DPRs). Overload of the proteasome system and reduced autophagy may contribute and/or cause this accumulation. (4) Impaired DNA repair: two recently identified ALS genes (see main text for details) work together in DNA repair, suggesting that impaired DNA repair could also contribute to ALS pathogenesis. (5) Mitochondrial dysfunction and oxidative stress: several ALS-related proteins (SOD1, TDP-43, C9orf72) can enter mitochondria and disrupt normal functioning, with increased formation of reactive oxygen species (ROS) as a consequence. (6) Oligodendrocyte dysfunction and degeneration, leading to reduced support for motor neurons. (7) Neuroinflammation: activated astrocytes and microglia secrete fewer neuroprotective factors and more toxic factors. (8) Defective axonal transport: several ALS-related mutations cause disorganization of the cytoskeletal proteins and disrupt axonal transport. (9) Defective vesicular transport: several ALS-related proteins (VABP, ALS2, CHMP2B, UNC13A) are involved in vesicular transport, suggesting that impaired vesicular transport contributes to ALS pathogenesis. (10) Excitotoxicity: loss of the astroglial glutamate transporter EAAT2 causes accumulation of extracellular glutamate, which causes excessive stimulation of glutamate receptors (e.g. AMPA receptors) and excessive calcium influx.
|