
- Title
-
Embryonic tissue differentiation is characterized by transitions in cell cycle dynamic-associated core promoter regulation
- Authors
- Wragg, J.W., Roos, L., Vucenovic, D., Cvetesic, N., Lenhard, B., Müller, F.
- Source
- Full text @ Nucleic Acids Res.
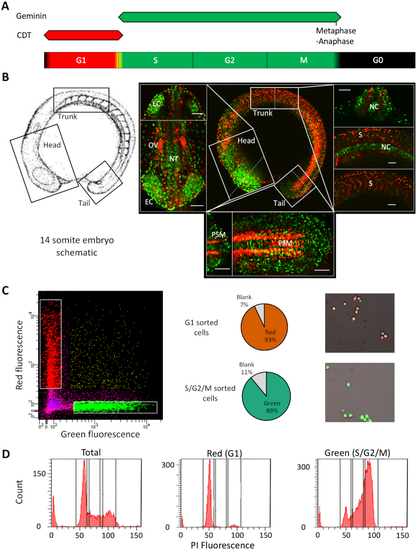
FACS mediated separation of cells by cell cycle stage in the developing embryo. ( |

Overview of CAGE samples. ( |

Classification of genes differentially expressed between G1 and S/G2/M segregated cells. ( |

Core promoter architecture. ( |
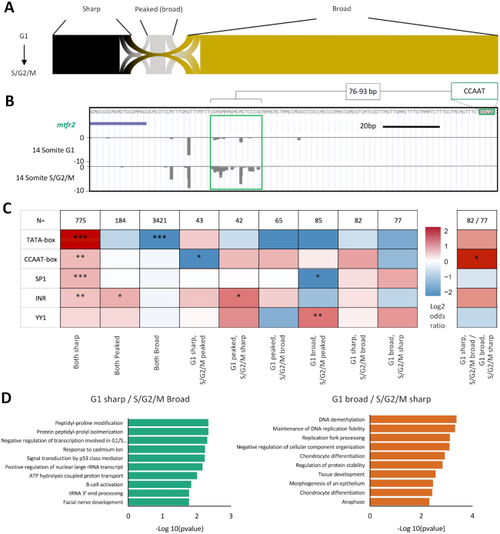
Core promoter shape transition. Promoter interquantile width (IQW) was measured and dominant TSSs assigned for consensus clusters (CCs) with at least 10 tpm expression in both the G1 and S/G2/M populations ( |

Alternative promoter usage. TPM values were determined for genomic regions identified in Nepal |
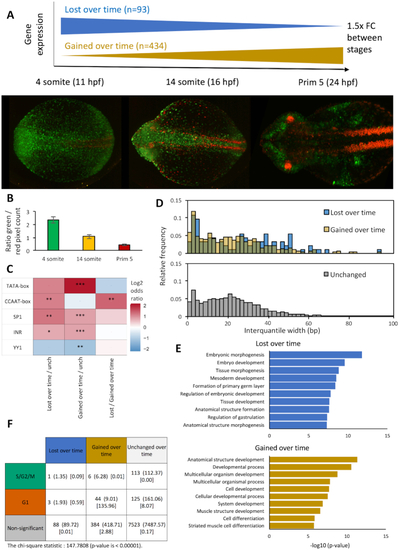
Genes with opposing temporal expression dynamics are marked by differential promoter motif utilization and TSS distribution. Published CAGE data from ( |