
- Title
-
Meta-Analysis of Grainyhead-Like Dependent Transcriptional Networks: A Roadmap for Identifying Novel Conserved Genetic Pathways
- Authors
- Mathiyalagan, N., Miles, L.B., Anderson, P.J., Wilanowski, T., Grills, B.L., McDonald, S.J., Keightley, M.C., Charzynska, A., Dabrowski, M., Dworkin, S.
- Source
- Full text @ Genes (Basel)

|
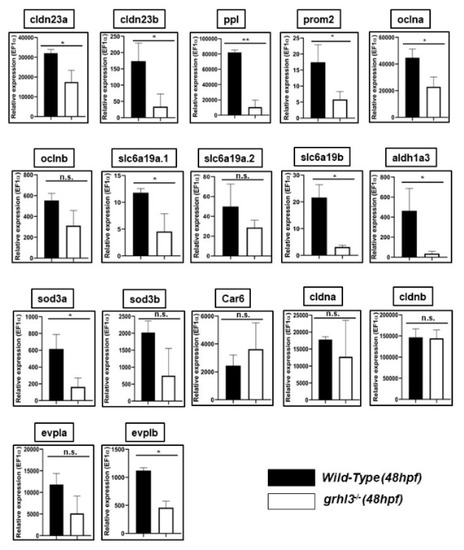
Q-RT-PCR analysis of zebrafish orthologue expression in 48 hours post-fertilisation (hpf) WT and |