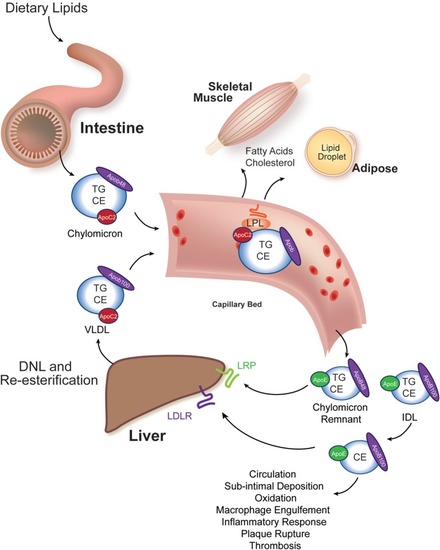
Intestinal and liver β-lipoprotein synthesis and vascular modification. Ingested lipids are hydrolyzed in the lumen of the intestine to absorbable species, such as free cholesterol, free fatty acids, and monoacylglycerol. These molecules are re-esterified in the enterocyte of the small intestine to triacylglycerol (TG), cholesteryl esters (CE), and phospholipids (not shown) and packaged into chylomicrons, whose signature coat protein in humans is Apob48 (one molecule per particle). This particle enters the vasculature and acquires an Apoc2 molecule from an HDL particle (not shown). Apoc2 is a required binding partner for lipoprotein lipase (LPL), an enzyme tethered to the apical surface of capillary bed endothelial cells in muscle and adipose tissues (20). LPL liberates free fatty acids for use by these tissues. The partially lipid-depleted chylomicron remnant is rapidly cleared by the liver through the action of Apoe-binding LRP receptors and Apob-binding low-density lipoprotein receptors (LDLR). The liver synthesizes very low-density lipoprotein (VLDL) particles from de novo lipogenesis-derived fatty acids and re-esterified fatty acids that reach the liver after adipocyte hydrolysis (and has relatively less CE in it). Human VLDL’s signature coat protein is Apob100. Following LPL-catalyzed lipid hydrolysis, VLDL remnants, intermediate density lipoprotein (IDL) particles, are either rapidly cleared by the liver or mature into LDL. LDL particles have a long circulating half-life, and they can deposit under vascular endothelial cells, undergo oxidation, and trigger an inflammatory atherosclerotic reaction with subsequent plaque rupture and thrombosis causing ischemia to the supplied tissue.
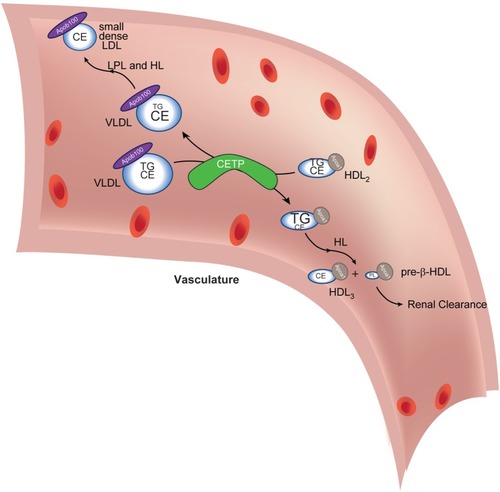
Cholesteryl ester transfer protein (CETP) in lipoprotein lipid exchange. Very low-density lipoprotein (VLDL) particles and HDL2 exchange triacylglycerol (TG) and cholesteryl esters (CE) in a reaction catalyzed by CETP. The depletion of TG and increase in VLDL CE (reflected in altered font sizes) coupled with lipoprotein lipase (LPL)- and hepatic lipase (HL)-mediated (further) depletion of TG (not shown) lead to the formation of small dense low-density lipoprotein (LDL), which is amenable to oxidative modification, a conversion central to driving subsequent atheromatous plaque formation. The transient increase in TG in HDL2 (reflected in increased font size) delivers a substrate for HL-mediated hydrolysis (as it passes through the liver capillaries). This reaction generates small HDL3 and pre-β-HDL, which contain scant amounts of phospholipids only. Pre-β-HDL is removed from the circulation via renal filtration. The net effect of CETP action, thus, is to cause maturation of VLDL into atherogenic, small, dense LDL and to decrease atheroprotective HDL concentration. Apoa1 is the signature coat protein of HDL.
Acknowledgments
This image is the copyrighted work of the attributed author or publisher, and
ZFIN has permission only to display this image to its users.
Additional permissions should be obtained from the applicable author or publisher of the image.
Full text @ Front Endocrinol (Lausanne)
Your Input Welcome
Thank you for submitting comments. Your input has been emailed to ZFIN curators who may contact you if
additional information is required.
Oops. Something went wrong. Please try again later.